Nevrofibromatose type 1
Nevrofibromatose type 1 er en medfødt sykdom som kan påvirke en rekke organer. Symptomene varierer derfor mye fra person til person. De fleste tegn på sykdommen viser seg allerede i barnealderen.

Sist oppdatert:
3. juni 2022
Det finnes to hovedtyper nevrofibromatose; type 1 (NF-1, også kalt von Recklinghausens sykdom) og type 2 (NF-2). Her finner du informasjon om nevrofibromatose type 1.
Hva er nevrofibromatose type 1(NF-1)?
Tilstanden skyldes genfeil på kromosom 17. Det er forskjellige typer genfeil som gjør at sykdommen kommer til uttrykk på ulik måte fra person til person.
NF-1 er en arvelig sykdom. Dersom en av foreldrene har tilstanden, er det 50 prosent risiko for at sykdomsgenet skal arves av hvert enkelt barn. Det er en autosomal dominant tilstand. Cirka halvparten av personer med NF-1 har friske foreldre. Dette skyldes at genfeilen i mange av tilfellene oppstår av seg selv, en såkalt spontan genmutasjon.
I en tysk studie fant man ett tilfelle per 3000 barn i seks-års alderen. Det er lik hyppighet i forekomst mellom kvinner og menn.
Symptomer
 Nevrofibromatose
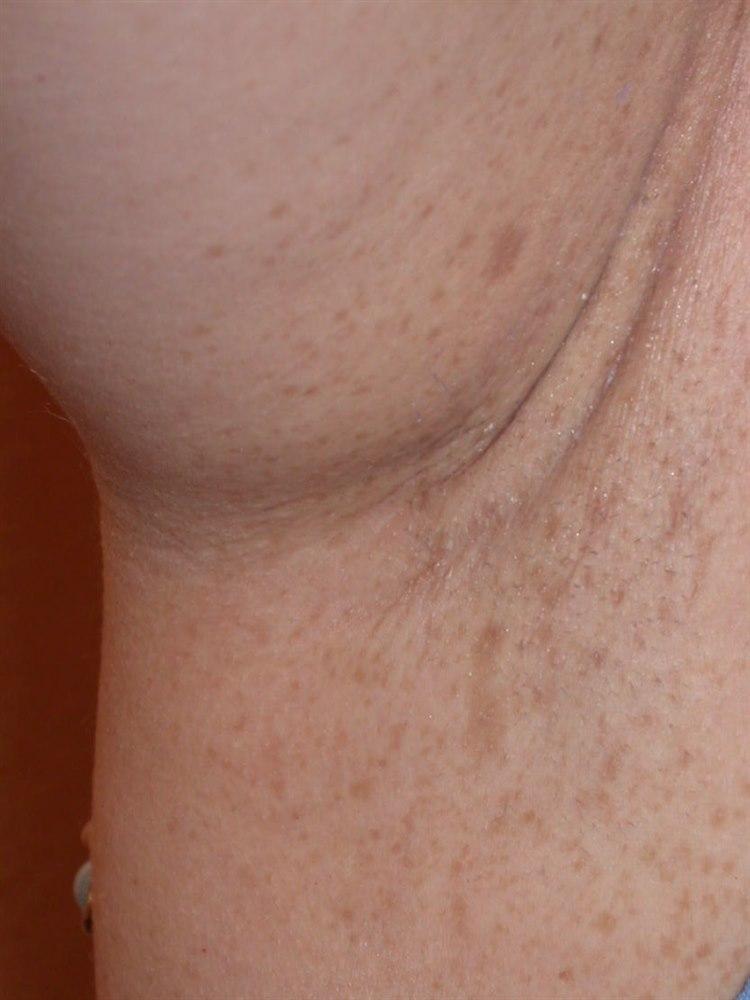
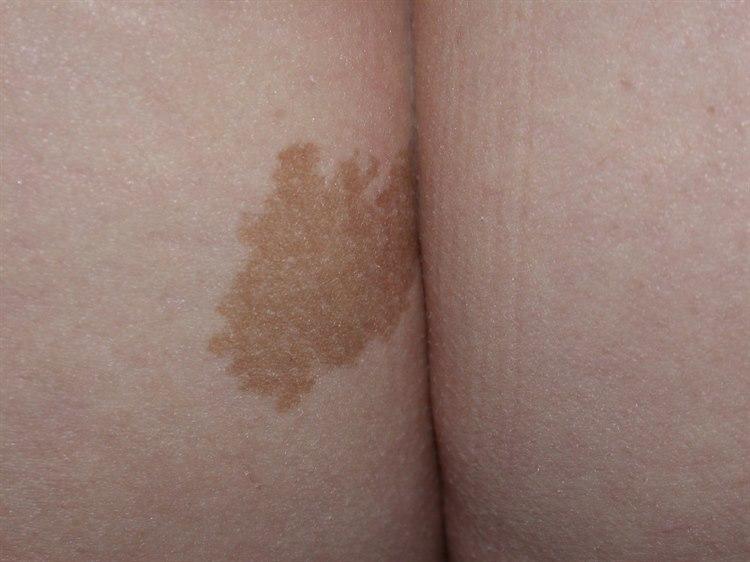
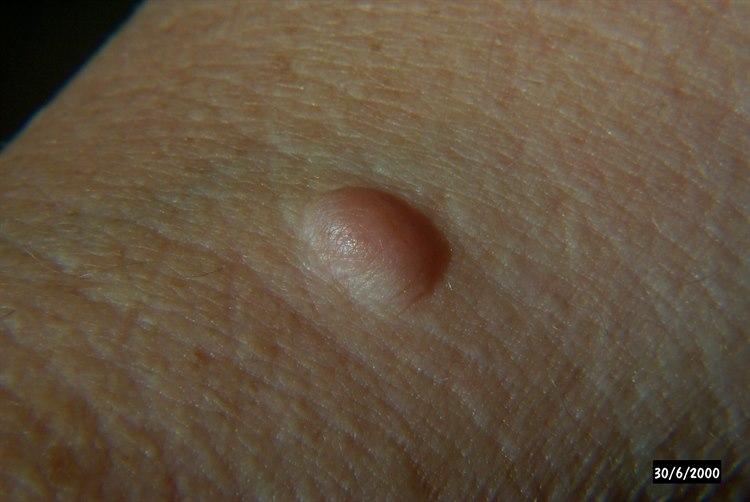
NevrofibromatoseI og med at mange ulike organer kan være involvert i sykdommen, vil symptomene kunne variere mye fra person til person. Symptomene utvikles som regel gradvis over tid. De fleste tegn på NF-1 viser seg allerede i barnealderen. Ved NF-1 er det vanlig med karakteristiske hudforandringer, såkalte café-au-lait-flekker. I tillegg er små svulster i huden vanlig, såkalte nevrofibromer. Ofte ses tette områder med fregner i lyske og armhule. Vekstforandringer i rørknokler er også vanlig.
Nevrofibromatose kan forårsake en rekke følgetilstander som høyt blodtrykk, lærevansker, atferdsproblemer, språkvansker og ADHD-symptomer. Psykisk og fysisk trettbarhet er vanlig og kan gi problemer med skolegang og arbeidsliv. Økt tilbøyelighet til beinbrudd som kan oppstå etter lette skader.
Diagnosen
Ved mistanke om nevrofibromatose er det viktig med en grundig legesjekk. Som regel vil det være hensiktsmessig med undersøkelse hos spesialist. Ulike røntgenundersøkelser, CT, MR, ultralyd osv. kan være aktuelle tilleggsundersøkelser. Genetisk testing er også mulig for å bekrefte diagnosen.
Behandling
Pasientene bør følges av nevrolog, nevrokirurg, øre-nese-halslege, øyelege, genetiker, fysioterapeut, sykepleier og fastlege. Det er vist at sentralisering av behandlingen bedrer prognosen.
Foreløpig finnes det ingen behandling som kan helbrede sykdommen eller stanse sykdomsutviklingen. Enkelte av svulstene som oppstår, kan fjernes ved vanlig kirurgisk metode eller ved såkalt strålekniv (gammakniv). Enkelte problemer kan lindres med medisiner. Hos barn med nevrofibromatose kan ulike varianter av habilitering være aktuelt.
Animasjon av strålekniv
Prognose
De første tegnene på sykdommen oppstår ofte tidlig, men de kan være uspesifikke. Ved målrettet legeundersøkelse bør man kunne stille diagnosen ved seks års alder. Forløpet av sykdommen er umulig å forutsi hos den enkelte.
Det er store variasjoner i hvor mye plager sykdommen gir. Enkelte kan leve tilnærmet normale liv og bli like gamle som befolkningen for øvrig. Andre vil ha ulike grader av problemer med helse, sosialt liv og arbeidsliv, og kan ha noe redusert levealder.
Det er viktig at personer med nevrofibromatose 1 blir fulgt opp med regelmessige undersøkelser.
Vil du vite mer
Dette dokumentet er basert på det profesjonelle dokumentet Nevrofibromatose type 1 . Referanselisten for dette dokumentet vises nedenfor
- Theos A, Korf BR. Pathophysiology of Neurofibromatosis Type 1. Ann Intern Med 2006; 144: 842-49. Annals of Internal Medicine
- Garg S, Green J, Leadbitter K, Emsley R, Lehtonen A, Evans DG, et al. Neurofibromatosis Type 1 and Autism Spectrum Disorder. Pediatrics 2013; 132: 1642-8. Pediatrics
- Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol 2005; 141: 71-4. pmid:15655144 PubMed
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988; 45(5):575-8. PubMed
- Garg S, Green J, Leadbitter K, et al. Neurofibromatsis Type 1 and autism spectrum disorder. Pediatrics 2013; 132: 1642-8. doi:10.1542/peds.2013-1868 DOI
- Kjærulff O, Wandall H. Sygdomsmekanismer og terapeutiske perspektiver ved neurofibromatose 1. Ugeskr Læger 2012; 174: 642. Ugeskrift for Læger
- Hamed Al-Farsi FA, Said Al-Alyani OB, Al-Kumzari A, Al-Saadi T. Systemic review and meta-analysis of the intellectual integrity of children with Neurofibromatosis type 1 (NF-1). World Neurosurg. 2021 Oct 11:S1878-8750(21)01578-3. PMID: 34648986. PubMed
- Krab LC, de Goede-Bolder A, Aarsen FK et al. Effect of Simvastatin on Cognitive Functioning in Children With Neurofibromatosis Type 1: A Randomized Controlled Trial . JAMA 2008; 300: 287-94. PubMed
- Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 2007; 61: 189-98. pmid:17387725 PubMed
- Ahlawat S, Blakeley JO, Langmead S, Belzberg AJ, Fayad LM. . Current status and recommendations for imaging in neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Skeletal Radiol 2020. pmid:31396668 PubMed