MPS VI (Maroteaux-Lamys sykdom)
Sist oppdatert:
11. aug. 2023
Innhold i artikkelen
Sammendrag
Definisjon:
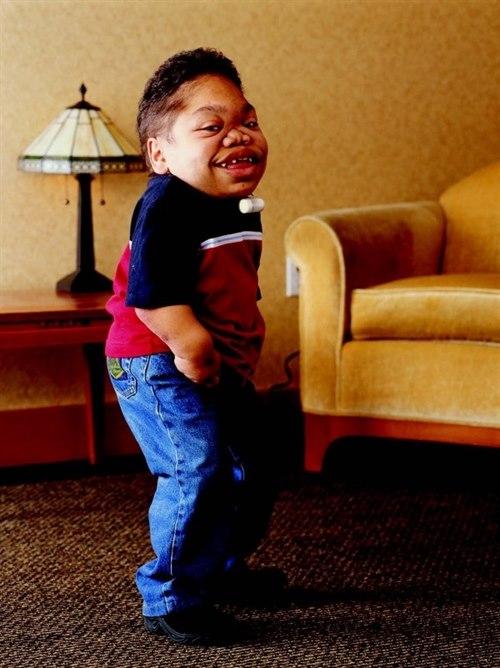
Arvelig (recessiv) tilstand som tilhører MPS-sykdommene (mukopolysakkaridose), en av de vanligste undergruppene av lysosymale sykdommer. Mutasjoner i genet ARSB påvirker produksjonen av enzymet N-acetylgalaktosaminsulfatase/arylsulfatase B og forårsaker mangelfull nedbrytning av dermatansulfat. Dette gir opphopning og avleiringer i kroppsvev som fører til forandringer i skjelettet og i hornhinnen i øyet, påvirker utseendet, og gir gradvis fremadskridende funksjonsnedsettelse
Forekomst:
1 av 400 000 levende fødte. Frambu kjenner til 3 tilfeller i Norge (2018)
Symptomer:
Stort spenn i hvor hardt sykdommen rammer. Utseendet kan være påfallende allerede ved fødselen, og stivhet i albueledd, hofteledd og knær utvikler seg vanligvis i løpet av de første leveårene. Barnets høydevekst er gjerne normal de første leveårene, men stagnerer ofte etter 6-8-årsalderen. Forventet slutthøyde ofte 110-140 cm. Potensielt økt infeksjonstendens. Kognitiv utvikling er forventet å være normal
Funn:
Relativt kort overkropp med utvikling av rykkskjevhet (torakolumbal gibbus. Nedsatt hørsel er vanlig. Forandringer i øyets hornhinne hos mange, ev. grønn stær eller forandringer i netthinnen. Nedsatt lungefunksjon og avleiringer i klaffer eller kransårer til hjertet forekommer. Kronisk diaré kan være et problem
Diagnostikk:
Klinisk undersøkelse, røntgenundersøkelse og sykdomshistorikk kan gi mistanke om tilstanden. Påvisning av glukosaminoglykaner (GAG) i urin gir sykdomsgruppen og bestemmelse av enzymaktivitet i celler fra blodprøve eller oppdyrkede celler (fibroblaster) fra hudprøve kan gi svaret. Gentesting er en annen (stadig vanligere) måte å stille diagnosen på. Diagnostikk før fødselen (prenatal) kan utføres ved fostervannsprøve, morkakeprøve eller genetisk preimplantasjonsdiagnostikk (PGD). PGD er bare mulig dersom familiens mutasjoner er identifisert
Behandling:
Årsaksrettet behandling i form av enzymerstatningsterapi (ERT): Galsulfase. Utredning og oppstart skjer på universitetssykehus. Enzymet tilføres intravenøst hver uke eller annenhver uke. Symptomrettet behandling av ev. komplikasjoner. Kirurgiske inngrep kan være/bli nødvendig. Internasjonale retningslinjer for oppfølging og behandling finner du her . Både barn og voksne med Maroteaux-Lamys sykdom bør ha ansvarsgruppe og individuell plan. Anestesi er komplisert, og krever ekstra kompetanse. Den gjennomsnittlige levealderen ved MPS VI er redusert, men avhenger av sykdomsuttrykket og behandlingen
Frambu sine nettsider om tilstanden ble besøkt 11.08.23
Legehandboka har kun sammendrag av Frambus omtale av diagnosen, teksten sammendraget er basert på finner du her:
Illustrasjoner
Bilder
Kompetansesenter
Nasjonalt senter for sjeldne diagnoser, enhet Frambu
- Frambu er et landsdekkende kompetansesenter for sjeldne og lite kjente funksjonshemninger
- Frambu er et statlig finansiert supplement til det ordinære hjelpeapparatet
- Frambu er en møteplass for familier og fagersoner
- Frambus tilbud er like mye til voksen som til barn - hele livsløpet
Nasjonalt senter for sjeldne diagnoser, enhet munnhelse
- Har et spesielt ansvar for sjeldne diagnoser som har betydning for munnhelse - bygge og spre kompetanse
- Alle som har en sjelden diagnose og en problemstilling knyttet til munnhelse eller funksjon, kan henvises til: Nasjonalt senter for sjeldne diagnoser, enhet munnhelse (tidligere TAKO-senteret)
- Består av et nasjonalt kompetansesenter for oral helse ved sjeldne diagnoser, samt en sykehustannklinikk som tar imot medisinske risikopasienter og personer med rus og psykiatri diagnose
- Lokalisert til Lovisenberg Diakonale Sykehus
- Enhet munnhelse har ikke oppfølgings- og behandlingsansvar, men bidrar med
- Råd og veiledning til lokale behandlere
- Utvikling av korte- og langsiktige behandlingsplaner
- Bistå med vurdering av tann- og kjeveutvikling, munnhelse og munnmotoriske funksjoner som spising og tale hos den enkelte
- Behandling kan utføres i spesielt kompliserte tilfeller som krever tverrfaglig kompetanse
- Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010 Apr 12;5:5. PMID: 20385007. www.ncbi.nlm.nih.gov